
Analysis of single-cell RNA-seq data, Part 2
Author: Marin Truchi
Analysis of single-cell RNA-seq data, Part 2
Biological interpretation of data
All analysis steps, from sequence alignment and counting to cluster annotation, data processing, normalization and integration, are carried out with the aim of obtaining the most complete and accurate description of the different cell populations composing the sequenced samples (see part 1). Cluster annotation can be a final goal, particularly in the case of projects seeking to map the resident cell types in adult human physiological tissues, fetal tissues or animal models. However, most single-cell RNA-seq experiments aim to answer biological questions that go beyond referencing cell populations and their respective markers. Indeed, these experiments are generally carried out to functionally characterize transcriptomic signatures induced by a treatment, pathology or genetic perturbation, and to follow their evolution during a differentiation pathway or development. These efforts to functionally interpret the observed biological mechanisms are based on statistical tests to describe variations in gene expression, and also rely on databases documenting the function of these genes, in order to hypothesize which regulatory pathways are modulated, from ligand-receptor interactions to transcription factor targets.
Differential expression analysis
Differential expression analysis (DEA) is the key step in quantifying variations in gene expression between cell populations or between conditions. Basically, for each gene, it consists in measuring the difference in its mean abundance between 2 groups and assessing whether this difference is statistically credible by means of a hypothesis test.
This analysis is particularly linked to the clustering step. Indeed, DEA first assesses the homogeneity and biological relevance of the specific transcriptomic signatures of each identified cluster. Conversely, clustering in a reduced-dimensional space free of sequencing depth bias and batch effects guarantees the relevance of the comparisons made by the DEA. These DEA include both comparisons between a cluster and the rest of the dataset to identify markers specific to a cell population, and comparisons between cells from different conditions within the same cluster to determine the modulations induced by each of them.
To be interpretable, these comparisons must be carried out on data that has been subjected to the minimum number of transformations. Indeed, although data normalization and integration are essential to reduce bias during clustering and for data representation, any corrections applied take them further away from their original measurements and are likely to artificially add variance for certain genes. The compromise generally found is to perform the DEA on data normalized to “CP10k” and then log transformed. As discussed in section 2.2.3, this method offers a number of advantages, including conservation of distances from the original measurements, reduction of sequencing depth differences, stabilization of variance, approximation of a normal distribution and expression of fold-change variations. While dozens of statistical tools have been proposed for performing DEA, it is appropriate to outline two typical cases, each requiring a specific type of approach.
Without biological replicates: Wilcoxon test on cell profiles
The first corresponds to experiments where only one or two samples per condition have been sequenced. In this case, the most commonly used method is to directly compare the expression profiles between the 2 sets of cells of interest using a Wilcoxon rank test. This non-parametric test consists in ranking in ascending order the normalized and log-transformed expression values of a gene among all the cells in the two sets combined. The sums of the ranks obtained for each group are then calculated, along with the probability of observing identical or more extreme values, on the assumption that there is no difference between the two groups being compared. As these pvalues are calculated for several thousand genes, a Bonferroni adjustment is applied to prevent the increase in type 1 error inherent in multiple testing.
This test, which has the advantage of being quick to perform and reliable, is nevertheless limited by the simplicity of the statistical model it proposes. In particular, it does not take into account any variability between samples. Indeed, when independent samples have been integrated, the cells are grouped together in each of the compared groups, without preserving this information. This neglect is likely to create situations where variations in the expression of certain genes are observed, whereas this effect is only carried by a fraction of the sequenced samples. Similarly, technical biases between samples that create variability are not taken into account, and create false positives. Finally, it has been shown that DEA methods based on counts measured in each cell are particularly biased for highly expressed genes, which tend to appear systematically differentially expressed.
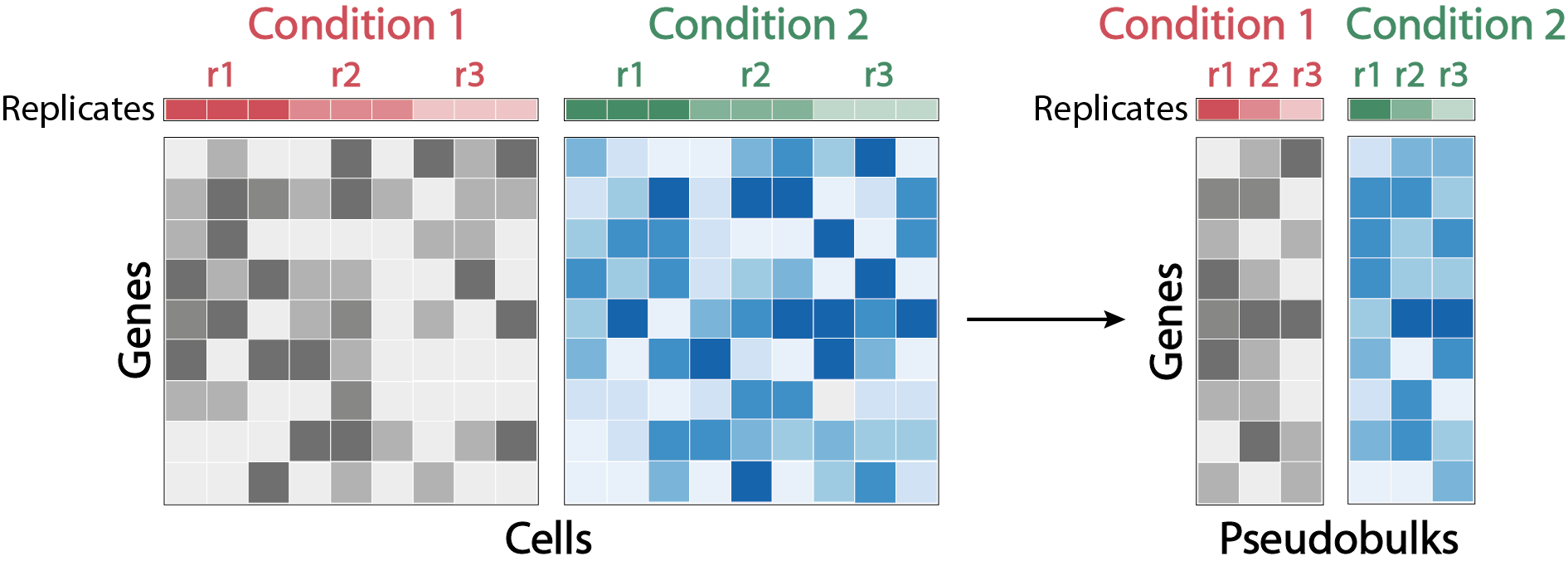
With biological replicates: DEA on “pseudobulks”.
When each entity being compared includes cells from several independent samples forming biological replicates, it is recommended to use a type of approach analogous to those used for DEA on “bulk” RNA-seq data. These approaches are recognized for their reliability, as their statistical model allows cofactors to be added to these parameters, and takes into account biological variability between replicates. However, due to the specificities of scRNA-seq data, notably the limited number of molecules quantified per cell, the application of these methods requires some adjustments.
The most common manipulation is to create “pseudo-bulk” samples by aggregating the counts of a population of cells from the same sample. Matrices of aggregated counts quantifying transcript expression per pseudo-bulk are processed with tools dedicated to DEA of RNA-seq data such as DESeq2, where pseudo-bulks corresponding to the same condition are considered in the model as biological replicates. Briefly, counts are normalized for each pseudo-bulk by the median of the ratios obtained by dividing the counts of each gene in the pseudo-bulk by the mean of the counts of the same gene in the whole dataset. The dispersion of each gene is then estimated by maximum likelihood, and related to the mean of its normalized counts. The observed dispersion of each gene is corrected according to its deviation from the dispersion expected by the model. This correction is increased when the number of samples is low, and genes whose observed dispersion greatly exceeds their expected dispersion are not corrected, so as not to add noise to the model. In this way, inter-sample variability is taken into account, preventing variations of technical origin from being detected as biological variations. Finally, each count is modeled according to a negative binomial probability distribution parameterized by its normalization coefficient and corrected dispersion. Based on these parameters, a Wald test is performed to test whether the hypothesis of equal expression between the 2 groups compared can be rejected. The associated pvalues are corrected for multiple testing using the Benjamini-Hochberg method. The amplitude of the variation is calculated by a log2 fold change, i.e. the log2 of the ratio between the mean of the modeled counts in the first group and those in the second group.
Although several comparative studies have demonstrated that the performance of this type of approach outperforms that of DEA methods based on the comparison of individual cell profiles, their robustness remains dependent on several factors. These include :
- the quality of pseudo-bulks, which depends on both the number of aggregated cells and the sequencing depth
- the quantity of pseudo-bulks, determined by the number of independent samples sequenced
- the amplitude and stability of the phenomenon causing the biological variability to be quantified.
Enrichment of gene sets and regulatory pathways
DEA produces transcriptomic signatures that define the identity of physiological cell types and states, or that are specific to a treatment, a pathology or a genetic modification. However, these signatures are not the final goal of the analysis. Rather, the aim is to interpret the identified signatures in order to hypothesize the involved functions and regulatory pathways. While some of the genes in these signatures are obvious indicators to an expert eye, or because of knowledge accumulated in the literature, most genes do not directly evoke these hypotheses. To overcome this, one of the most common approaches is to query databases listing the roles associated with these different genes, in order to identify possible recurrent functions.
This type of analysis, known as gene set enrichment (GSEA), is based on the principle that genes involved in the same mechanism will tend to be co-expressed. At the statistical level, this involves testing whether certain functions are over-represented in DEA-derived signatures compared with signatures obtained by random sampling. The genesets associated with molecular functions, regulatory pathways, cellular compartments, pathologies or responses to a stimulus are multiple, since they originate from different initiatives. These genesets are themselves stored in databases such as MsigDB, which contains a total of 33196 genesets, divided into several large collections. These include the genesets collection of Gene Ontology and the collection of genesets related to canonical regulatory pathways such as KEGG. A gene set enrichment analysis produces a table where each geneset listed in the queried collections is associated with an enrichment score, a statistical value corrected for multiple testing that estimates the significance of this score, and the list of genes that contribute to the enrichment. This approach has a number of limitations, the main one being the interpretability of gene sets that appear enriched. Indeed, depending on the collections queried, the terms defining the enriched sets may be redundant, imprecise or irrelevant to the studied biological system. As a result, the lists produced by enrichment analyses require selection, which is likely to bias the results.
A complementary approach to gene set enrichment analyses is to focus not on the genes involved in regulatory mechanisms, but directly on genes that are impacted downstream of these signaling pathways. PROGENy proposes to compare the expression profiles of cell populations with signatures established from public data documenting the transcriptional impact of perturbations to 11 major signaling pathways. An activity score for each of these pathways is calculated in each population according to the expression level of genes specific to these 11 signatures. The advantage of this approach is that it is less supervised and produces more biologically interpretable results. Nevertheless, the 11 pathways referenced are rather relevant to the study of cancers, and are not sufficient for the study of certain systems where other pathways are involved. Finally, as with gene set enrichment analysis, performance on single-cell RNA-seq data is generally inferior to that on bulk RNA-seq data, for which these approaches were developed, due to the difference in sequencing depth.
Inference of intercellular communication
In addition to gene set enrichment analyses, other approaches can be used to infer hypotheses about the regulatory pathways at the origin of the transcriptomic signatures observed for a cell population. Some of these approaches focus on potential ligand-receptor interactions capable of initiating a signaling cascade leading to modulation of gene expression.
CellphoneDB is a database which, by cross-referencing public proteomic interaction data, provides a catalog of genes encoding receptor subunits and ligands. Based on the expression levels of these different genes in the identified cell populations in a dataset, CellphoneDB establishes communication patterns between populations expressing ligands and those expressing receptor subunits. In a similar concept, NicheNet proposes a more complex model. Indeed, in addition to cross-referencing ligand-receptor interaction data, the model is also based on protein-protein interaction data and on regulatory data between transcription factors and their target genes. In this way, complete signalling and regulatory pathways are reconstituted, by weighting the interactions between each of their elements by a score, proportional to the reliability and abundance of their evidence. The final model synthesizes this information into a table assigning a potential regulatory score between each ligand listed and target genes modulated at the terminal level of the process. The analysis carried out with NicheNet consists in defining a set of genes likely to be induced following a signaling cascade initiated by an interaction between a ligand and a receptor, as well as a set of populations likely to express this ligand and the receptor population of this signal. By comparing the set of genes of interest with the a priori regulatory model, NicheNet generates a score for the potential activity of ligands capable of inducing transcription of these target genes in the “receiver” population. The credibility of this inferred activity is assessed according to the amplitude of the score calculated, the level of expression of the ligands expressed by the “sender” populations, and the level of receptors specific to these ligands in the “receiver” population. Despite its greater complexity, NicheNet remains limited. Like CellphoneDB, it compares a model of interactions based on proteomic data with scRNA-seq expression data, while the two modalities may show limited correlation. It is conceivable that these models will be improved in the near future by the addition of information from highly resolutive spatial transcriptomic approaches, capable of adding an histological dimension to ligand-receptor interaction predictions.
Gene regulatory network inference
To better understand the regulatory processes involved in differential gene regulation across cell types or conditions, gene regulatory networks (GRN) can be inferred from scRNA-seq data. Based on the assumption that members of a particular GRN are co-expressed, tools such as WGCNA or GRNBoost2 aim to find correlations between gene expression profiles and build co-expression modules. SCENIC was the first adaptation to scRNA-seq data to improve the model by generating cell-type-specific GRN, linking the expression profiles of genes coding for transcription factors (TF) to other genes, weighting the edges within each GRN with a probability of enrichment of TF binding motifs on the promoter regions of co-expressed genes. However, relying exclusively on transcriptomic data is not suitable for GRN inference, as the mRNA level of a TF-encoding gene may be a poor estimate of the TF’s functional protein. In addition, this model does not take into account an important factor in what constitutes a GRN, which is the state of chromatin accessibility. Indeed, binding of TF to cis-regulatory elements (promoters, enhancers, silencers, distals) requires unpacking the particular DNA sequence. In this context, gene expression data must be complemented by binding data from Chip-seq or CUT&Tag assays, as well as chromatin accessibility data from ATAC-seq or DNAse-seq assays.
The combination of scATAC-seq and scRNA-seq is probably the most mature multiomics approach for linking changes in chromatin accessibility to gene transcription. However, the analysis of these multimodal data is not straightforward, as it depends on several factors. Firstly, the two profiling can be performed simultaneously in the same cell or independently, which may require an integration process. Even if they were measured in the same cell, the underlying kinetics of the two levels of regulation are not necessarily simultaneous. In any case, GRN inference is limited by the sparsity of single-cell measurements. Then, GRN modeling based on both chromatin accessibility and transcriptome profiling depends on the mathematical assumptions adopted to represent their interaction (linear or non-linear), as well as on the chosen statistical framework. Furthermore, inferring the regulatory activity of a TF on a target gene via a potential TF binding event relies both on querying databases of TF binding motifs and on defining the chromosomal distance between the binding event and the target gene. Consequently, a multitude of GRN modeling tools implementing these different possibilities have been developed (listed here). However, none of them can overcome some of the limitations of GRN inference, such as taking into account the 3D structure of the genome or TFs without known binding motifs. Finally, benchmarking the performances of existing tools cannot be based on comparison with the ground truth.
Trajectory inference based on similarity of expression profiles
A final type of analysis frequently carried out on scRNA-seq data consists in attempting to recreate a pathway of differentiation linking several cell populations. This analysis, summarized by the term trajectory inference, is based on the assumption that this process is continuous and is reflected at transcriptomic level by a progressive evolution of expression profiles. Although sequencing represents a snapshot of cellular states, making it impossible to follow this evolution for a single cell over time, it does assume that sequenced profiles can correspond to different stages in the differentiation process.
This dynamic can then be reconstructed by organizing the cells according to the similarity of their expression profiles, starting with cells that correspond to the presumed initial state, to those that potentially constitute a differentiated final state. The position of each cell along this trajectory is expressed in terms of a quantitative variable called pseudo-time, which corresponds to the transcriptomic distance separating this cell from its initial state, and reflects its progress. Modeling this trajectory depends mainly on assumptions about the lineage relationships of cell populations and the complexity of the expected topology. Indeed, some models require the definition of an initial population to determine the orientation of the trajectory, while others estimate it from the data. Similarly, the shape of the trajectory is either constrained by predefining the number of initial states, branch points and final states, or left flexible.
A large number of trajectory inference tools have been proposed, each using their own combination of statistical methods and their own starting hypotheses. However, these methods share a common structure, which is articulated in 2 steps. They begin with a dimension reduction step to capture the variables relevant to describing the position of cells on the trajectory. A graph linking each cell by edges of length proportional to the distances in the reduced-dimension space is then constructed. From this graph, a trajectory is constructed according to different strategies, depending in particular on the definition of a starting point.
The choice and use of trajectory inference methods remains a controversial subject, due to the limitations of this type of approach. Comparison between methods is difficult, as their performance depends above all on the biological system studied, in which the ground truth is not always known. Indeed, the performance of most methods is presented using hematopoiesis as an example, which in no way guarantees that this performance will be maintained in other systems where the lineage between cell populations is unknown. Moreover, several trajectories can be found depending on the parameter settings, leaving the user free to potentially choose the one that best corresponds to his a priori idea of the differentiation pathways in his dataset.
RNA velocity analysis
The main alternative for revealing transcriptional dynamics from scRNA-seq data is to model the cycle of RNA transcription, splicing and degradation. This so-called “RNA velocity” analysis is based on distinguishing the reads corresponding to unspliced immature transcripts from those corresponding to mature transcripts spliced during alignment. The two types of reads are quantified separately so as to obtain 2 count matrices for the same cells and genes, one with unspliced transcripts and the other with spliced transcripts. From these data, the evolution of mature and immature RNA quantities over time is modeled by a system of differential equations. The amount of unspliced RNA depends on both the rate of gene transcription and the rate of splicing, while the amount of spliced RNA depends on the rate of immature RNA splicing and the rate of mature RNA degradation.
Ideally, the phase portrait representing the expression of the immature transcript of a gene as a function of the expression of the mature transcript in each cell resembles two arcs of circles linking two stable states, the first around the origin and the second at the opposite end. The first arc corresponds to the transcription phase, when the quantity of unspliced RNA grows faster than that of spliced RNA due to a relative delay in maturation, until it reaches a stable state where the two quantities are equivalent due to the termination of transcription. The second arc represents the repression phase, where both quantities progressively decrease in a staggered manner. Since model parameters such as transcription, splicing and degradation rates cannot be calculated by regular measurements in different cell populations, they are estimated using an expectation-maximization algorithm. Briefly, this algorithm involves assigning each cell a time point, minimizing the distance from the expected trajectory, and a state (stable, undergoing transcription or undergoing repression), in order to calculate the expectation of the likelihood function of the model parameters. In a second step, the parameters that maximize this likelihood function are used to update the model. These steps are repeated iteratively until convergence is reached. A velocity vector is calculated for each gene in each cell, taking the differences between the ratio of immature to mature RNA observed and that expected in a steady state. The individual gene vectors are then combined for each cell to estimate their future state, which is assumed to be already represented by one of its nearest neighbors. A similarity matrix between the velocity vector calculated for a cell and the potential displacement vectors between this cell and its neighbors is used to determine which of the latter is most likely to represent this future state. In this way, the cells are ordered according to their assumed anteriority relationships, giving the trajectory an orientation.
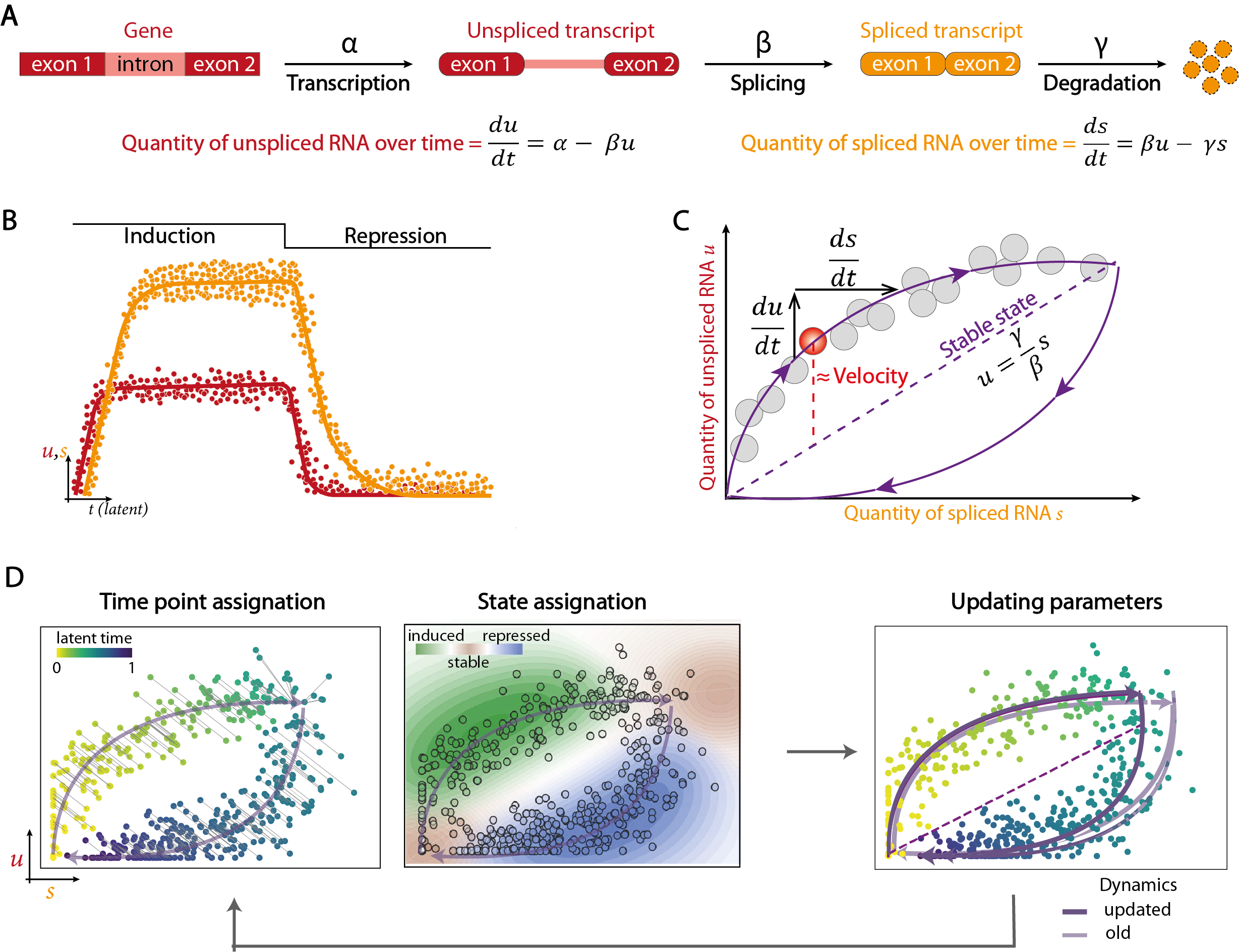
Although trajectory determination by RNA velocity is less biased than by similarity of expression profiles, this approach also has a number of limitations. Firstly, the differential equation model assumes that transcription, splicing and degradation rates are constant, whereas these events are subject to stochasticity (bursting) and complex regulatory mechanisms (epigenetics, alternative splicing, post-transcriptional modifications) that induce heterogeneity. Furthermore, model parameterization relies heavily on the reading of phase portraits representing the evolution of the ratio between unspliced and spliced RNA. However, due to the complexity of regulatory mechanisms and the low capture capacity of single-cell molecules, few genes present the ideal phase portrait presented above. As a result, parameter estimation remains largely noisy. Finally, quantification of transcripts by reading the 3’ ends, typical of protocols based on cell isolation in droplets, is not ideal for identifying immature RNA. With this type of technology, quantification of unspliced and spliced transcripts is highly dependent on the tools used for this task. Whole transcript sequencing approaches, offering greater depth and better definition of intronic regions, therefore appear more suitable for this type of analysis.
Each of the trajectory inference approaches has its own limitations. Those based on similarity of expression profiles generally require a priori knowledge to determine the direction and origin of the trajectory, while those based on RNA velocity are sensitive to noise and difficult to interpret. To take advantage of the complementarity between these 2 approaches and limit their biases, CellRank proposes to combine the robustness of cell organization based on similarity of expression profiles with trajectory orientation information indicated by RNA velocity. However, not all the biases inherent in both approaches are eliminated, and the analysis still relies on a major assumption concerning sequencing. To follow the evolution of a differentiation between several cell populations, all intermediate stages must be captured. In some cases, this requires the sequencing of several samples, possibly at different time points. The maximum time interval between each point should then be consistent with transcriptional kinetics. What’s more, the integration of several samples may add noise to the analysis if the technical variability between samples is significant.
Errors to avoid in the second part of the analysis
- Ignore potential biological replicates in differential analysis
- Use trajectory inference/RNA velocity analysis in the absence of potential intermediate stages.
- Over-interpret the results of the various tools used to infer biological hypotheses from the data, without taking into account the limitations of their models.
Bibliography
Nguyen, H.C.T., Baik, B., Yoon, S. et al. Benchmarking integration of single-cell differential expression. Nat Commun 14, 1570 (2023).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Ogbeide, S., Giannese, F., Mincarelli, L. & Macaulay, I. C. Into the multiverse: advances in single-cell multiomic profiling. Trends Genet. 38, 831–843 (2022).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Weiler, P., Van den Berge, K., Street, K. & Tiberi, S. A Guide to Trajectory Inference and RNA Velocity. Methods Mol. Biol. Clifton NJ 2584, 269–292 (2023).
Badia-i-Mompel, P., Wessels, L., Müller-Dott, S. et al. Gene regulatory network inference in the era of single-cell multi-omics. Nat Rev Genet (2023).
Lange, M., Bergen, V., Klein, M. et al. CellRank for directed single-cell fate mapping. Nat Methods 19, 159–170 (2022).